The polymerase chain reaction laboratory technique is used in a variety of applications to make copies of a specific DNA sequence. This lesson describes how a PCR reaction works, what it accomplishes and its basic requirements for success. Examples of interpreting results are given. PCR's strengths, weaknesses and applications to plant biotechnology are explained.
 Lesson Navigation Tips: Lesson Navigation Tips:- Click on ’Animations’ button found to the left in order to view the animation which supplements this lesson. You can also click on the animation icon within the text. - Click once on figures to see enlarged versions. - Click once on words in color to bring up their definitions. |
In 1983 a scientist by the name of Kary Mullis was driving along a Californian mountain road late one night and formulated a revolutionary way to make laboratory copies of DNA molecules (Saiki et al. 1985, Mullis 1990). The resulting polymerase chain reaction, or PCR for short, probably first became known to the general American public in its forensics use during the 1995 O.J. Simpson trial. In general PCR is sort of like making a genetic photocopy of a section of a chromosome.
This laboratory technique is modeled after a living cell’s natural ability to replicate (make copies of) DNA during normal cell cycles. Every living cell makes a duplicate copy of each chromosome during the early stage of cell division known as interphase.
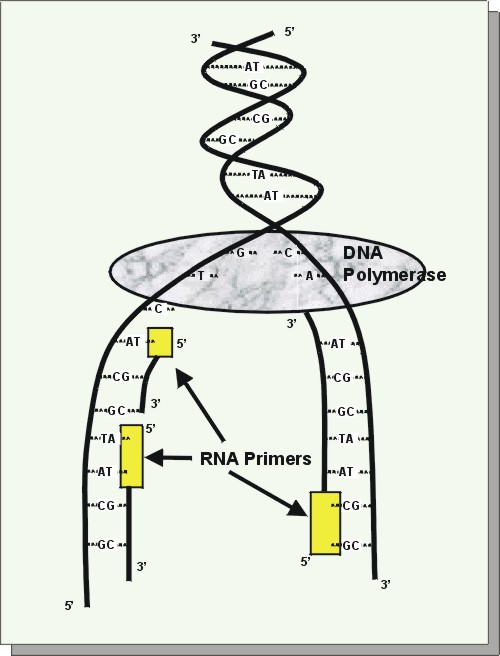 |
| DNA replication: As the 2 DNA strands of a chromosome pull apart, RNA primers bind. Then DNA polymerase completes the copying process. |
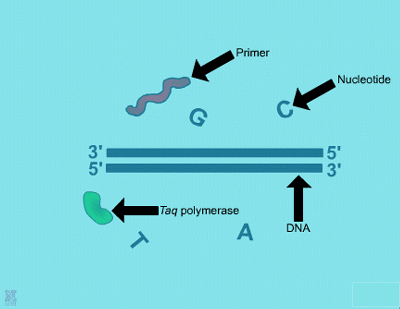
Let’s now take a look at what the name 'Polymerase Chain Reaction' describes in order to better understand the details of the process. 'Polymerase' is chosen because PCR makes use of a DNA polymerase enzyme for constructing new DNA strands, just like in a living cell. 'Chain Reaction' is also used because this technique involves repeating different heating and cooling cycles over and over again, as many as 35 or more times. Each complete cycle doubles the amount of DNA present, so in just 20 cycles there are over one million (220) copies of that specific segment of DNA . For PCR there are five chemical components needed, including a DNA template, DNA polymerase enzyme, primers, nucleotides and reaction buffer. These are described here in detail.
1. The DNA template is that particular DNA sequence which you want copied.
2. There are two requirements for a suitable DNA polymerase enzyme for PCR. First, one is needed that has a good activity rate around 75°C. Second, it should be able to withstand temperatures of 95-100°C so that more enzyme does not have to be added at the beginning of each new cycle. The first to be discovered with these characteristics was Taq polymerase, isolated from Thermus aquaticus, a thermophilic eubacterium found in hot springs (Chien et al. 1976). Naturally coming from a hot environment, it does not easily denature in the hot temperatures required in PCR; plus it has a good efficiency, able to add 60 base pairs/sec at 70°C. Like all other DNA polymerases, Taq cannot begin DNA replication without the addition of a starting primer.
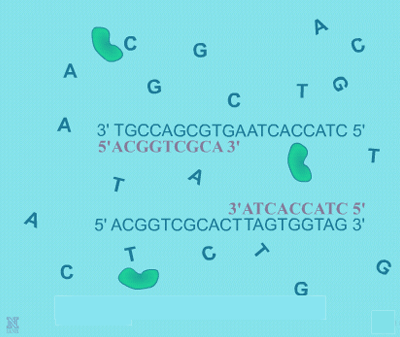
3. Primers are short oligonucleotides of DNA, usually around 8-60 base pairs in length. They can be random sequences if the project’s goal is for general genomic studies. However, if the purpose is to amplify a certain section of DNA in the genome, such as a known gene, then primers of specific sequences must be used.
4. The four different deoxyribonucleotide triphosphates (dNTPs), adenine (A), guanine (G), cytosine (C), and thymine (T) are needed to provide the building blocks for DNA replication. DNA polymerase will add each complementary base to the new growing DNA strand according to the original strand’s sequence following normal A-T and C-G pairings.
5. Finally, a reaction buffer is used to provide a stable pH. It may also contain magnesium chloride, if not, then MgCl2 must be added separately. Mg2+ plays a vital role in the PCR reaction, acting as a co-factor for Taq polymerase and thereby influencing enzyme activity.
 |
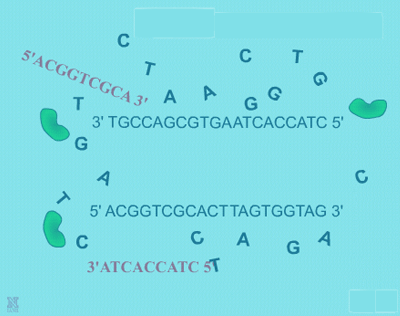
| In general, a single PCR run will undergo 25-35 cycles. The first step for a single cycle is the denaturation step, in which the double-stranded DNA template molecule is made single-stranded. The temperature for this step is typically in the range of 95-100°C, near boiling. The high heat breaks the hydrogen bonds between the strands (Figure: Denaturation). The second step is a primer annealing step in which the primers bind to complementary sequences in the single-stranded DNA template. The general temperature range for this step is 45-55°C. | |||
 |
 |
||
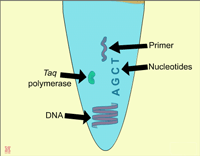
| Denaturation: 1. This shows the beginning of the first step of PCR, the denaturation step. Shown are the chemical components, double-stranded DNA template, nucleotide bases, primers and DNA polymerase enzyme molecules. | 2. In this step, the PCR mixture is heated causing the hydrogen bonds to break and the DNA to become single stranded. | ||
|
|||
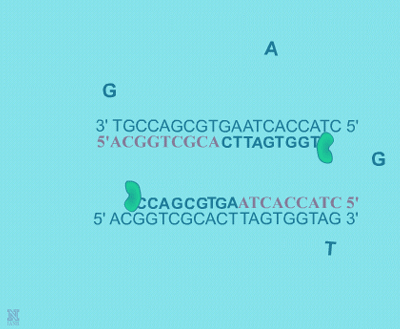 |
 |
||
| Extension: The final PCR step is when the DNA polymerase enzyme (ie. Taq polymerase) hooks new bases to the primer, extending a new complementary piece of DNA. | Completion of the final step and the first cycle of PCR, resulting in a doubling of the amount of DNA template present. | ||
| Finally, the DNA extension step is when the DNA polymerase enzyme (i.e. Taq polymerase) adds bases complementary to the template to the bound primers (Figure: Extension). The chemical mixture is heated to around 75°C and the newly synthesizing DNA strand is extended to the end of the template area to be copied. The completion of this step is also the completion of one cycle. For cycle 2, the denaturation, annealing, and extension steps are repeated again. This time, though there will be twice as many DNA template molecules compared to what there was at the beginning of cycle 1. In other words, copies are being made of the original template and of the copies made in the previous cycle. Therefore, if everything is working correctly, at the end of a cycle there is double the amount of that particular DNA sequence as what was found at the beginning of the cycle. |
|||
|
|||
Now that a complete PCR reaction has been completed, we need to be able to see the results. To do this, a sample of the PCR mixture is loaded into an agarose gel for electrophoresis. An agarose gel looks something like a thin block of jello. It contains a matrix of pores which enables it to separate DNA fragments based on their sizes. See lesson on Electrophoresis: How Scientist observe fragments of DNA
 |
| Electrophoresis: A gel electrophoresis set-up with the power supply on the upper-left and agarose gel with DNA and loading dye on the right. Photo by Brian Bolles, Colorado State University. |
Here is an actual photograph
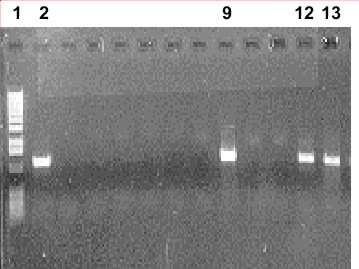 |
| Photograph: A photograph of an agarose gel showing results of a PCR experiment. Photo by Deana Namuth, Colorado State University. |
A couple of PCR’s strengths are that it is a simple procedure to set up and run, and requires only small amounts of starting DNA samples. It is now possible to get enough DNA to conduct a PCR analysis from merely a paper hole punch size of leaf tissue. Even just one human hair contains enough DNA, making PCR useful in forensics. Another advantage of PCR is that it is a very quick procedure. In as little as 2 hours, millions of copies of DNA can be made. Automation and robotic assistance are now available which allow the processing of many samples in a very short time. In a context such as plant breeding, this would mean that many individual plants can be sampled by simply collecting a small leaf tissue. They could then be 'PCRed' to screen for the presence of a particular gene of interest. Only those testing positive would be kept for the next plant breeding stage. For example, an automated PCR would make it simple to keep only those plants with a transgene after a backcross. See Lesson: Real Time PCR - Some Basic Principles.
There are some drawbacks of using PCR that one should be aware of as well. First, if you are wanting to amplify a specific gene, you will need some knowledge of the gene’s DNA sequence in order to properly design some primers. In this case, specific primers are used so that amplification of only the gene takes place, rather than random areas of the chromosome. There may also be a need to optimize concentrations of each chemical component. For example changing the amount of DNA template, MgCl2 and Taq polymerase can affect both the quantity and quality of bands produced. Some studies have shown that even the brand of Taq polymerase can affect results (Holden et al. 2003). Likewise the temperature cycles may need adjusted.
When PCR was originally developed, DNA fragments in the range of 100 - 500 bp could be copied. Now there are modifications which allow successful PCR for DNA fragments up to 5 kb, but any larger and the reaction becomes less efficient (Barnes 1994; Cheng et al. 1994). There are reports of long extension PCR with fragments of as large as 42 kb being amplified, but these types of reactions are not yet routine (Barnes 1994; Cheng et al. 1994). Therefore, PCR is currently limited to copying only parts of genes or other DNA clones.
PCR can be a tool used in plant breeding programs. For example, instead of screening for insect resistance by scoring infested plants, a breeder can score based on the presence of a marker which is linked to the insect resistance gene. This reduces the chance of environmental effects which could inhibit symptom expression, indicating a plant is resistant, when in reality it is susceptible. In another scenerio, PCR can be used
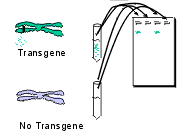 |
| Application: Since complementary primers have been designed to bind specifically to the transgene, PCR will be successful only in plants which are truly transgenic. At the end of PCR a sample from each are run through a gel. No band will be seen with non-transgenic plants. |
To briefly highlight the topics of this lesson, remember that PCR is a relatively easy lab technique which amplifies the amount of DNA present, much like living cells do in the beginning stages of a cell cycle. There are 5 chemical components of a PCR reaction: a DNA template, a DNA polymerase, primers, nucleotides, and a buffer. The 3 temperature steps for one cycle are the denaturation, primer annealing and extension steps. There are now many variations and uses of PCR ranging from forensics to genomic studies to identifying transgenic crops. PCR can be very useful for determining whether or not a plant contains a transgene.
Barnes W (1994) PCR amplification of up to 35-kb DNA with high fidelity and high yield from lambda bacteriophage templates. Proc. Natl. Acad. Sci. USA 91:2216-2220.
Cheng S, Fickler C, Barnes W and Higuchi R (1994) Effective amplification of long targets from cloned inserts and human genomic DNA. Proc Natl. Acad. Sci. USA 91:5695-5699.
Chien A, Edgar DB, Trela JM (1976) Deoxyribonucleic acid polymerase from the extreme thermophile Thermus aquaticus. J. Bacteriol 127: 1550-1557.
Holden MJ, Blasic JR, Bussjaeger L, Kao C, Shokere LA, Kendall DC, Freese L, and Jenkins GR (2003). Evaluation of extraction methodologies for corn kernel (Zea mays) DNA for detection of trace amounts of biotechnology-derived DNA. Journal of Agricultural and Food Chemistry (51)9:2468-2474.
Mullis K (1990) The unusual origin of the polymerase chain reaction. Scientific American 262:56-65.
Saiki RK, Scharf S, Faloona F, Mullis KB, Horn GT, Erlich HA, Arnheim N (1985) Enzymatic amplification of B-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 230:1350-1354.